染色体の進化
染色体の本数は種によって違っていて、進化の過程で、融合したり、分裂したりしてきたと考えられる。現在、いくつかの種では、染色体単位でゲノム配列が決定されているので、異種間の染色体を比較して、染色体が、どう変化してきたのか、見てみようと思った。
ゲノム配列がまだ解読されてなかった1996年のレビュー
人類の染色体進化研究の現状
https://doi.org/10.1537/ase.104.355
の要約には、
ヒトの染色体進化は, かつてはバンド・パターンを指標とした近縁霊長類との比較により研究されてきた。 しかし最近では, 遺伝子工学的手法によるDNAレベルでの染色体比較研究が主流となっている。
とある。第一文は、多分、染色体の分染法に基づく染色パターンの比較のことを言ってるのだろう。1968年に、Q分染法が発表され、1971年に、G分染法が考案されたらしい。第二文で言及されてる手法は、上レビューのテーマであるFISH(fluorescence in situ hybridization)法のことと思う。当時は、こうして得られる限られた情報を使って、染色体が、どのように変化してきたか推測していたらしい。
レビュー中の図4を見ると、ヒトと、6種の哺乳類の染色体の対応関係を示している。2020年代の現在は、ここに挙げられている動物は、染色体単位で、塩基配列が決定されているので、ずっと詳細な情報を、数時間で得ることができる。更に、近縁の哺乳類だけでなく、もっと遠いニワトリやトカゲとの比較もできる。
そういう論文は沢山出ているけど、読むのが面倒なので、自分で適当に実験していく。
例えば、ヒトとアカゲザル(学名Macaca mulatta、日本では外来種とされるが、ニホンザルと交雑するらしい)の比較をしてみる。やることは、それぞれの各染色体同士のアラインメントの作成。アラインメントには、LASTZを使った。
LASTZ
https://github.com/lastz/lastz
make lastz_32でビルドされるLASTZ_32というのも入れておく。最新のバージョンは、1.04.15だったので、それを使用。
ゲノム配列は、UCSCの以下のURLからダウンロード。
Sequence and Annotation Downloads
https://hgdownload.soe.ucsc.edu/downloads.html
ヒトは、hg38が最新。アカゲザル(一般名はRhesus macaque)は、rheMac10が最新。hg38.faとかrheMac10.faは、複数の配列を含んでるので、一ファイル一配列に分割しておく。染色体以外の配列chrUn*とか、chr*randomとかあるけど、それらは除外しておく。ミトコンドリアの配列もあるけど、不要なら消しておく。それぞれの配列ファイルを、hg38とrheMac10ディレクトリに置いたとして、以下のようなコマンドで、アラインメントを作成した。
spc1=hg38
spc2=rheMac10
for f in `ls ${spc1}`;do
for f2 in `ls ${spc2}`;do
lastz_32 ${spc1}/$f ${spc2}/$f2 --notransition --step=20 --nogapped --format=general:nmismatch,name1,strand1,start1,end1,size1,name2,strand2,start2,end2,size2 > ${spc1}_vs_${spc2}/${spc1}_`basename $f .fa`_${spc2}_`basename $f2 .fa`.out
done
doneこれで、mismatchは許すが、gapは許さない(ラフな)アラインメントができる。一致しない塩基同士がある時、gapになるか、mismatchになるかは、penalty次第だけど。
このアラインメントを可視化するために、ドットプロットを作る。以下のような実装を作った
import matplotlib.pyplot as plt
from matplotlib.ticker import MaxNLocator
import os,sys
#-- dot plot for single ".out" file
def dotplot(filepath , threshold=1000):
Nunit = 1000000 #-- per Mb
dat = []
size1,size2 = 0,0
name1,name2="",""
for line in open(filepath):
line = line.strip()
if len(line)==0 or line[0]=="#":continue
ls = line.split()
if len(ls)<10:continue
name1,name2 = ls[1],ls[6]
s0,e0 = int(ls[3]),int(ls[4])
s1,e1 = int(ls[8]),int(ls[9])
size1 = int(ls[5])
size2 = int(ls[10])
if ls[2]=="-":s0,e0=size1-e0,size1-s0
if ls[7]=="-":s1,e1=size2-e1,size2-s1
if ls[2]==ls[7]:
dat.append( (s0,e0,s1,e1,+1) )
else:
dat.append( (s0,e0,s1,e1,-1) )
if len(dat)==0:return
fig, ax = plt.subplots()
dat.sort(key=lambda x:x[-1])
ax.set_xlim(0 , size1/Nunit)
ax.set_ylim(0 , size2/Nunit)
ax.set_xlabel("{0} (Mb)".format(name1))
ax.set_ylabel("{0} (Mb)".format(name2))
ax.xaxis.set_major_locator(MaxNLocator(integer=True))
ax.yaxis.set_major_locator(MaxNLocator(integer=True))
#ax.set_aspect('equal')
for s0,e0,s1,e1,dd in dat:
if e0-s0 < threshold:continue
if dd==+1:
ax.plot([s0/Nunit,e0/Nunit],[s1/Nunit,e1/Nunit],color="red")
else:
ax.plot([s0/Nunit,e0/Nunit],[e1/Nunit,s1/Nunit],color="blue")
plt.savefig("{0}.png".format(filepath))
plt.close()
#plt.show()
def make_dotplot(dirpath , threshold=1000):
for f in os.listdir(dirpath):
dotplot(os.path.join(dirpath,f) , threshold)
if __name__=="__main__":
make_dotplot(sys.argv[1])
thresholdは、ヒトとチンパンジーのように近縁種の場合は、似た配列が沢山あるのと、プロット量が多いので、1000でいいけど、近縁でない種の場合は、もっと小さい数値(例えば、0)にしないと、対応関係が見えない。
冒頭の総説を見ると、ヒトの2番染色体は、ニホンザルの9番と15番染色体に対応してるとある。染色体番号の付け方が違うようで分かりづらいが、アカゲザルでは、多分、chr12,chr13と対応してる。ヒトの2番染色体は、他の大型類人猿が持つ2本の染色体が融合して生じたと考えられている。UCSCにあるチンパンジーゲノム配列では、chr2Aとchr2Bという名前になってるけど、チンパンジーのchr2Bとアカゲザルのchr13が対応する染色体らしい。
ヒトの2番染色体は、ブタでも2本の染色体に対応してるので、ブタとサルのMRCA(Most Recent Common Ancestor)に於いても、2本の染色体だったのだろうと思われる。ブタ(susScr11)のchr3とchr15は、チンパンジーのchr2A,chr2Bと、それぞれ相同性がある。
冒頭のレビュー図4によると、ニホンザルの7,13番染色体は、ヒト染色体では、14,15番染色体と20,22番染色体に対応している。ニホンザルの7,13番染色体は、アカゲザルでは、chr7とchr10に相当すると思われる。例えば、アカゲザルの7番染色体とヒト14,15番染色体間のドットプロットは以下のようになった。
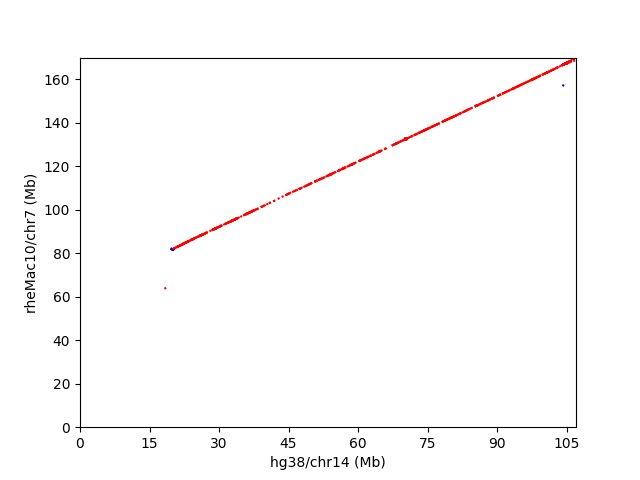
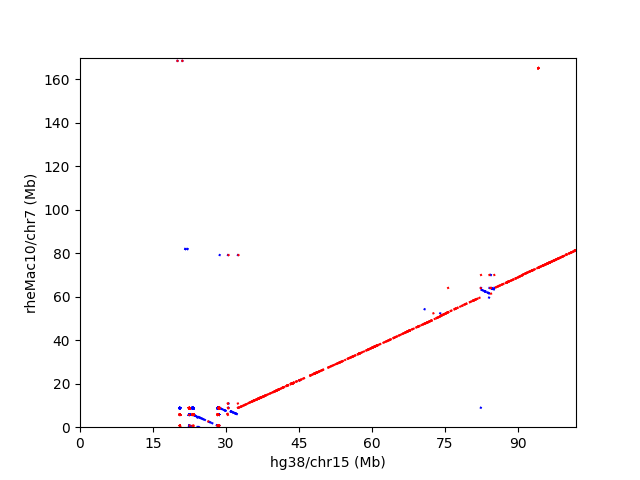
赤い部分は傾きが正で、同一strand。青い部分は傾きが負で、strandが逆になってる部分を表す。ゴリラの17番染色体とヒトの5番染色体のドットプロットは、以下のようになっている。
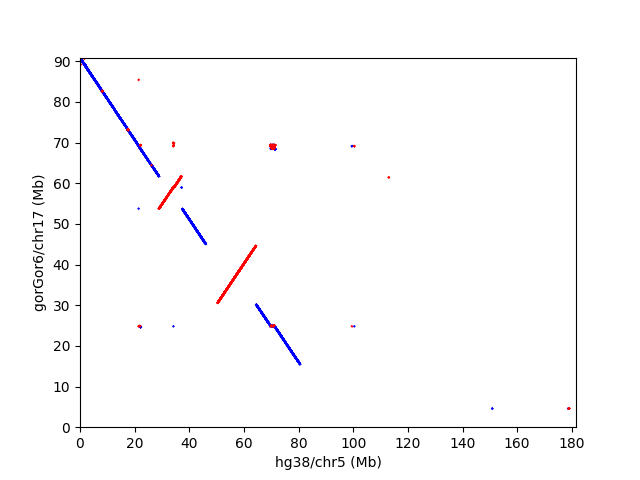
ドットプロットは全部作ったけど、数が多いので他は省略。
ドットプロットを目で見ると、染色体が、どう再編成したかアタリが付く。gapなしのアラインメントは、アラインメントされない領域が多すぎるし、また複数箇所にマップされることも多いので、以下のようなコマンドで、なるべく広範囲の精密なアラインメントを行う。
s1=(1 2 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22)
s2=(1 12 13 2 5 6 4 3 8 15 9 14 11 17 7 7 20 16 18 19 10 3 10)
for i in "${!s1[@]}"; do
lastz_32 hg38/chr"${s1[$i]}".fa rheMac10/chr"${s2[$i]}".fa --chain --gfextend --gapped --step=20 --format=maf > hg38_vs_rheMac10/pairs_$i.maf
doneこっちの方が、計算時間は長いので、gapなしのアラインメントで、染色体対応関係のアタリを付けておくのは、計算時間の短縮のために有益。
以前に、大型類人猿間で、同じオプションで、ゲノム間アラインメントを作って、ミスマッチ率を計算したけど、ゴリラの染色体再編成を考慮したり、以前は、2A染色体と2B染色体を結合して、ヒト2番染色体と比較してたのを、それぞれ別々にやって計算し直したのが以下の表。ついでに、指標を追加した。
| 種1 | 種2 | 染色体 | アライン長(bp) | mm数(bp) | gap数(bp) | 延べアライン長(bp) | mm率(%) | umm数(bp) | 一意gap数(bp) | 一意アライン長(bp) | umm率(%) | dmm数(bp) | dmm率(%) | cmm数 |
| hg38 | panPan3 | 常,X | 2678735571 | 41576138 | 31189401 | 2717358723 | 1.53 | 6758108 | 4406603 | 503091302 | 1.34 | 3215657 | 0.12 | 2456798 |
| hg38 | panTro6 | 常,X | 2699947183 | 44404643 | 33778773 | 2751026993 | 1.61 | 6483380 | 4236391 | 484545618 | 1.34 | 3925816 | 0.14 | 2971884 |
| panTro6 | panPan3 | 常,X | 2686365237 | 16904614 | 16975214 | 2726251998 | 0.62 | 3021545 | 2565225 | 688400052 | 0.44 | 1775208 | 0.07 | 1243923 |
| 種1 | 種2 | 染色体 | アライン長(bp) | mm数(bp) | gap数(bp) | 延べアライン長(bp) | mm率(%) | umm数(bp) | 一意gap数(bp) | 一意アライン長(bp) | umm率(%) | dmm数(bp) | dmm率(%) | cmm数 |
| hg38 | gorGor6 | 常,X | 2626582175 | 48739956 | 33858948 | 2652779980 | 1.84 | 8802977 | 5360986 | 511789105 | 1.72 | 3440313 | 0.13 | 2693551 |
| panTro6 | gorGor6 | 常,X | 2565732892 | 47670060 | 31548473 | 2590652189 | 1.84 | 9707027 | 5614943 | 566282817 | 1.71 | 3297893 | 0.13 | 2606152 |
| panPan3 | gorGor6 | 常,X | 2533630218 | 47149463 | 31197440 | 2559477278 | 1.84 | 9437399 | 5462824 | 551406301 | 1.71 | 3284119 | 0.13 | 2591233 |
| 種1 | 種2 | 染色体 | アライン長(bp) | mm数(bp) | gap数(bp) | 延べアライン長(bp) | mm率(%) | umm数(bp) | 一意gap数(bp) | 一意アライン長(bp) | umm率(%) | dmm数(bp) | dmm率(%) | cmm数 |
| hg38 | ponAbe3 | 常,X | 2588246045 | 93093484 | 57653867 | 2625495453 | 3.55 | 15351101 | 8323644 | 460620083 | 3.33 | 8341840 | 0.32 | 6876853 |
| panTro6 | ponAbe3 | 常,X | 2487178819 | 89772146 | 53999989 | 2522771809 | 3.56 | 15455544 | 8213412 | 461475178 | 3.35 | 8063035 | 0.32 | 6647953 |
| panPan3 | ponAbe3 | 常,X | 2482052601 | 89520144 | 53594491 | 2518426197 | 3.55 | 15632330 | 8300557 | 466525516 | 3.35 | 7960499 | 0.32 | 6599212 |
| gorGor6 | ponAbe3 | 常,X | 2498049196 | 89470029 | 52566349 | 2528024398 | 3.54 | 16011592 | 8387101 | 476595807 | 3.36 | 7725178 | 0.31 | 6452555 |
アライン長:アラインメントされた配列の長さの合計(オーバーラップしてる場合、重複カウントなし)
mm数:アラインメントのmismatch数
gap数:アラインメントのgap数
延べアライン長:アラインメントされた配列の長さの合計(オーバーラップしてる場合、重複してカウント)
mm率:mm数/延べアライン長
umm数:一意にmapされた領域のmismatch数
一意gap数:一意にmapされた領域のgap数
一意アライン長:一意にmapされた領域の長さ合計
umm率:一意mm数/一意アライン長
dmm数:2bp連続でmismatchがある箇所の個数。dmmは私が作った造語
dmm率:dmm数/延べアライン長
cmm数:連続してmismatchがある領域の個数
SNV(single nucleotide variant)が完全にランダムな場所に落ちるなら、dmm率は、mm率の2乗に近い値になるはずだけど、そうはなってなくて、近縁種ほど、乖離が大きい。表にはないけど、一意にアラインされた領域でも、ほぼ同じなので、複数箇所にアラインメントを許してることに起因するartifactとかではなさそうに思える。
cmm数は、ミスマッチが、3つ以上続く領域が沢山ある可能性を排除するために測定したもの。1+(dmm数/cmm数)で、2つ以上ミスマッチが続く領域の平均長が分かる。
SNVがランダムに落ちるわけでないのは、染色体ごとのmm率のバラツキを漫然と眺めてても何となく分かるけど、単一の指標があった方が理解しやすい。染色体ごとに、dmm率を計算してみると、各染色体でも、SNVの入りやすい領域と、そうでない領域が存在してるらしいことが分かる。dmm率/(mm率の2乗)を計算すると、ゲノム単位と染色体単位で、それほど差はない。
アカゲザル(rheMac10)と大型類人猿の間で、同様に比較すると、以下のようになってた。
| 種1 | 種2 | 染色体 | アライン長(bp) | mm数(bp) | gap数(bp) | 延べアライン長(bp) | mm率(%) | umm数(bp) | 一意gap数(bp) | 一意アライン長(bp) | umm率(%) | dmm数(bp) | dmm率(%) | cmm数 |
| hg38 | rheMac10 | 常 | 2285895996 | 156162266 | 141741288 | 2338797775 | 6.68 | 29669259 | 22429338 | 468373954 | 6.33 | 20826992 | 0.89 | 16735778 |
| panTro6 | rheMac10 | 常 | 2004548986 | 137987342 | 124474482 | 2053284974 | 6.72 | 26716942 | 20069769 | 419761766 | 6.36 | 18546755 | 0.90 | 14883475 |
| panPan3 | rheMac10 | 常 | 2004762223 | 137329895 | 123850821 | 2051694693 | 6.69 | 26673714 | 20026072 | 419686539 | 6.36 | 18326651 | 0.89 | 14743807 |
| gorGor6 | rheMac10 | 常 | 1927929371 | 131494040 | 117016510 | 1969649600 | 6.68 | 25746189 | 19215645 | 403458141 | 6.38 | 17380587 | 0.88 | 14026091 |
| ponAbe3 | rheMac10 | 常 | 2138375821 | 147872816 | 133646631 | 2190175646 | 6.75 | 30374712 | 22559771 | 474878246 | 6.40 | 19909886 | 0.91 | 15991518 |
ミスマッチ率が、ほぼ等しいので、大型類人猿で、SNV獲得速度に差は、殆どなかっただろうと思う。もっと遡るために、新世界ザルであるコモンマーモセット(calJac4)との比較を行う。
| 種1 | 種2 | 染色体 | アライン長(bp) | mm数(bp) | gap数(bp) | 延べアライン長(bp) | mm率(%) | umm数(bp) | 一意gap数(bp) | 一意アライン長(bp) | umm率(%) | dmm数(bp) | dmm率(%) | cmm数 |
| rheMac10 | calJac4 | 常 | 1959286889 | 231664102 | 252003127 | 2023901958 | 11.45 | 53629550 | 51874175 | 481270165 | 11.14 | 43922606 | 2.17 | 33976695 |
| hg38 | calJac4 | 常 | 1879486713 | 209382873 | 227920796 | 1934888846 | 10.82 | 47766735 | 46318223 | 453739763 | 10.53 | 38202814 | 1.97 | 29807236 |
| panTro6 | calJac4 | 常 | 1823134238 | 203598006 | 220695032 | 1878129858 | 10.84 | 46686827 | 45009615 | 442227433 | 10.56 | 37174828 | 1.98 | 29001488 |
| panPan3 | calJac4 | 常 | 1814251852 | 202007402 | 218818327 | 1867886607 | 10.81 | 46362676 | 44768625 | 439965764 | 10.54 | 36788720 | 1.97 | 28721677 |
| gorGor6 | calJac4 | 常 | 1794640696 | 198573615 | 213752368 | 1844481101 | 10.77 | 45878734 | 44005838 | 436158798 | 10.52 | 35847561 | 1.94 | 28066581 |
| ponAbe3 | calJac4 | 常 | 1959775525 | 218832288 | 237063460 | 2017994375 | 10.84 | 51280402 | 49461086 | 486281052 | 10.55 | 39960867 | 1.98 | 31190420 |
アカゲザルとコモンマーモセットの間のミスマッチ率は、大型類人猿とコモンマーモセットの間のミスマッチ率より高そうなので、アカゲザルは、大型類人猿より、一年あたりのSNV得速度が早い可能性が推測される。アカゲザルは、大型類人猿より性成熟が早く、寿命もやや短いようなので、一世代あたりのSNV獲得量自体は、同水準なのかもしれない。
染色体対応関係とコマンドは、以下のような感じ。
s1=(1 1 1 2 2 3 3 3 4 5 6 7 8 8 9 10 10 11 12 13 13 14 15 15 16 16 17 18 19 20 21 22)
s2=(18 19 7 14 6 15 17 21 3 2 4 8 13 16 1 12 7 11 9 1 5 10 10 6 12 20 5 13 22 5 21 1)
for i in "${!s1[@]}"; do
lastz_32 hg38/chr"${s1[$i]}".fa calJac4/chr"${s2[$i]}".fa --chain --gfextend --gapped --step=20 --format=maf > hg38_vs_calJac4/pairs_$i.maf
dones1=(1 1 1 2 2 2 3 3 3 4 5 6 7 7 8 8 9 9 10 10 11 12 13 14 15 16 17 17 18 19 20 20)
s2=(18 19 7 15 17 21 2 21 8 4 3 2 10 6 13 16 12 7 1 5 9 6 14 11 1 5 1 5 13 22 12 20)
for i in "${!s1[@]}"; do
lastz_32 rheMac10/chr"${s1[$i]}".fa calJac4/chr"${s2[$i]}".fa --chain --gfextend --gapped --step=20 --format=maf > rheMac10_vs_calJac4/pairs_$i.maf
done
まぁ、とりあえず、1996年の総説にあったような染色体進化の追試は、大分簡単にできるようになった(結論)。
現在、サル目の分類は、以下のようになっているらしい。上記で比較したサル目の動物は、全部、真猿に含まれる。
---- サル目 ------------------ 曲鼻亜目 -------- キツネザル下目
| |
| └-- ロリス下目
|
|
└------- 直鼻亜目 -------- メガネザル下目
|
|
└-- 真猿下目 ---------- 新世界ザル
|
|
└----- 旧世界ザル(オナガザル上科)
|
|
└-- 類人猿(ヒト上科) ----------- 小型類人猿(テナガザル科)
|
└----- 大型類人猿(ヒト科) 系統が遠くなるにつれて、アラインメントされる長さも減ってる。こういう比較が、どれくらいまで使えるのか調べようと思って、脊索動物の幅広い種で、gapなしのアラインメントした。ドットプロットを見てると、系統的に遠い種では、大きな相同領域があることは期待できないけど、もう少し定量的に把握するために、coverageを計算した。
種1と種2のゲノムをgapなしで、アラインメントして、種1のゲノム配列の内、種2のどこかにマッピングされた領域の長さが、種1のゲノム全長の何%に相当するかを被覆率1、同様に、種1のどこかにマッピングされた領域の長さが、種2のゲノム全長の何%に相当するかを被覆率2として集計したのが以下の表。
ゲノム配列は、基本的に、UCSCにリンクがあるのを使ったけど、ハリモグラはなかったし、ジャイアントパンダは古かったので、以下でゲット
mTacAcu1.pri
https://www.ncbi.nlm.nih.gov/assembly/GCF_015852505.1/
Ailuropoda melanoleuca (giant panda)
https://www.ncbi.nlm.nih.gov/assembly/GCF_002007445.2
| 種1 | 種2 | 被覆率1(%) | 被覆率2(%) | 通名1 | 通名2 | |
| hg38 | panTro6 | 42.95 | 51.67 | ヒト | チンパンジー | |
| hg38 | panPan3 | 42.49 | 64.33 | ヒト | ボノボ | |
| hg38 | gorGor6 | 41.76 | 53.61 | ヒト | ゴリラ | |
| hg38 | ponAbe3 | 41.60 | 38.70 | ヒト | オランウータン | |
| hg38 | rheMac10 | 39.24 | 45.24 | ヒト | アカゲザル | |
| hg38 | nomLeu3 | 39.95 | 40.57 | ヒト | テナガザル | |
| hg38 | calJac4 | 30.47 | 37.20 | ヒト | マーモセット | |
| hg38 | oryCun2 | 4.35 | 6.90 | ヒト | ウサギ | |
| hg38 | mm39 | 2.67 | 3.87 | ヒト | マウス | |
| hg38 | rn7 | 2.62 | 5.93 | ヒト | ラット | |
| hg38 | canFam6 | 6.14 | 15.01 | ヒト | イヌ | |
| hg38 | felCat9 | 6.61 | 7.20 | ヒト | ネコ | |
| hg38 | ailMel3 | 4.88 | 6.19 | ヒト | パンダ | |
| hg38 | bosTau9 | 5.54 | 9.58 | ヒト | 牛 | |
| hg38 | susScr11 | 5.74 | 6.73 | ヒト | ブタ | |
| hg38 | monDom5 | 1.33 | 1.09 | ヒト | オポッサム | |
| hg38 | tacAcu1 | 0.76 | 1.36 | ヒト | ハリモグラ | |
| hg38 | ornAna2 | 0.24 | 1.42 | ヒト | カモノハシ | |
| hg38 | galGal6 | 0.49 | 2.46 | ヒト | ニワトリ | |
| hg38 | xenLae2 | 0.30 | 0.48 | ヒト | アフリカツメガエル | |
| hg38 | danRer11 | 0.21 | 2.10 | ヒト | ゼブラフィッシュ | |
| hg38 | oryLat2 | 0.15 | 0.91 | ヒト | メダカ | |
| hg38 | ci3 | 0.01 | 0.29 | ヒト | ユウレイボヤ | |
| panTro6 | panPan3 | 46.23 | 52.19 | チンパンジー | ボノボ | |
| felCat9 | oryCun2 | 4.06 | 4.78 | ネコ | ウサギ | |
| felCat9 | canFam6 | 19.62 | 44.92 | ネコ | イヌ | |
| canFam6 | ailMel3 | 20.46 | 11.49 | イヌ | パンダ | |
| bosTau9 | susScr11 | 11.73 | 12.15 | ウシ | ブタ | |
| bosTau9 | canFam6 | 7.21 | 13.95 | ウシ | イヌ | |
| susScr11 | felCat9 | 8.66 | 10.36 | ブタ | ネコ | |
| ornAna2 | tacAcu1 | 28.69 | 8.67 | カモノハシ | ハリモグラ | |
| ornAna2 | monDom5 | 1.64 | 0.10 | カモノハシ | オポッサム | |
| mm39 | rn7 | 23.94 | 31.21 | マウス | ラット | |
| rn7 | felCat9 | 2.73 | 3.73 | ラット | ネコ | |
| rn7 | monDom5 | 1.09 | 0.55 | ラット | オポッサム | |
| galGal6 | ornAna2 | 0.33 | 0.34 | ニワトリ | カモノハシ | |
| galGal6 | anoCar2 | 0.81 | 0.19 | ニワトリ | グリーンアノール | |
| fr3 | oryLat2 | 5.05 | 2.74 | フグ | メダカ | |
| fr3 | gasAcu1 | 7.58 | 6.22 | フグ | トゲウオ | |
| oryLat2 | gasAcu1 | 3.45 | 9.97 | メダカ | トゲウオ | |
| danRer11 | fr3 | 0.64 | 2.83 | ゼブラフィッシュ | フグ |
ここで算出されたcoverageの絶対的な数字自体に、さほどの意味はないけど、系統的に近い種同士だと、被覆率が高くなる傾向は見て取れ、目安程度にはなる。
ヒトと他のサル目は被覆率が高いし、同じネコ目に属するイヌとネコ、ネズミ目に属するマウスとラットも被覆率が高い。ウシとブタは、鯨偶蹄目に分類されている。パンダもネコ目に属し、ヒトよりは、イヌやネコに近そう。パンダが、どれくらいクマか算出したかったけど、染色体の配列まで決定されたクマのゲノムがなかった。哺乳類の中でも、単孔目のカモノハシやハリモグラくらいまで行くと、ヒトと比較した時の被覆率は大分低い。
ニワトリと爬虫類のグリーンアノールは、あんまり似てないっぽいけど、現行の爬虫類の中で、トカゲやヘビを含む有鱗目は、鳥類から遠い系統で、最も近いのは、ワニ目とされている。最近は、以下のような系統関係が支持されてるらしい。
------------ 有鱗目(ヘビ、トカゲ、ミミズトカゲ)
|
----------------|
| |
| ------------ ムカシトカゲ目
---- 爬虫類 -------|
| ------------ カメ目
| |
----------------|
| --------- ワニ目
------------|
--------- 鳥類この系統関係が正しければ、多分、ワニとトカゲも、ゲノムは、それほど似てないと思われる。現在、利用可能なワニゲノムは、アメリカアリゲーターのもので、染色体スケールで配列が決定されてなかったので、今回は保留。単純なゲノム全体の相同性で分類するなら、ヘビ・トカゲ。ムカシトカゲと、カメ・ワニ・鳥に分けるほうが理に適ってるのかもしれない。とりあえず、ニワトリとグリーンアノールの比較結果も妥当ではあるんだろう。
鳥は特徴的な外見(嘴、羽毛、翼など)で区別できるので、ワニがトカゲと鳥に近いか質問して、鳥と答える人はいないだろうし、何の知識もない3歳児に、色んな生物種を提示して鳥か鳥でないかクイズを実施しても、あんまり間違えなさそうに思える(コウモリを鳥と答える子供はいるかもしれないけど)。なので、鳥に固有の配列を探索したという研究もある。
ゼブラフィッシュは、コイ目で、骨鰾上目に含まれる。フグはフグ目、メダカはダツ目で、トゲウオ(≠トビウオ、トビウオはダツ目)はトゲウオ目で、フグ目とダツ目とトゲウオ目は、棘鰭上目に分類されている。
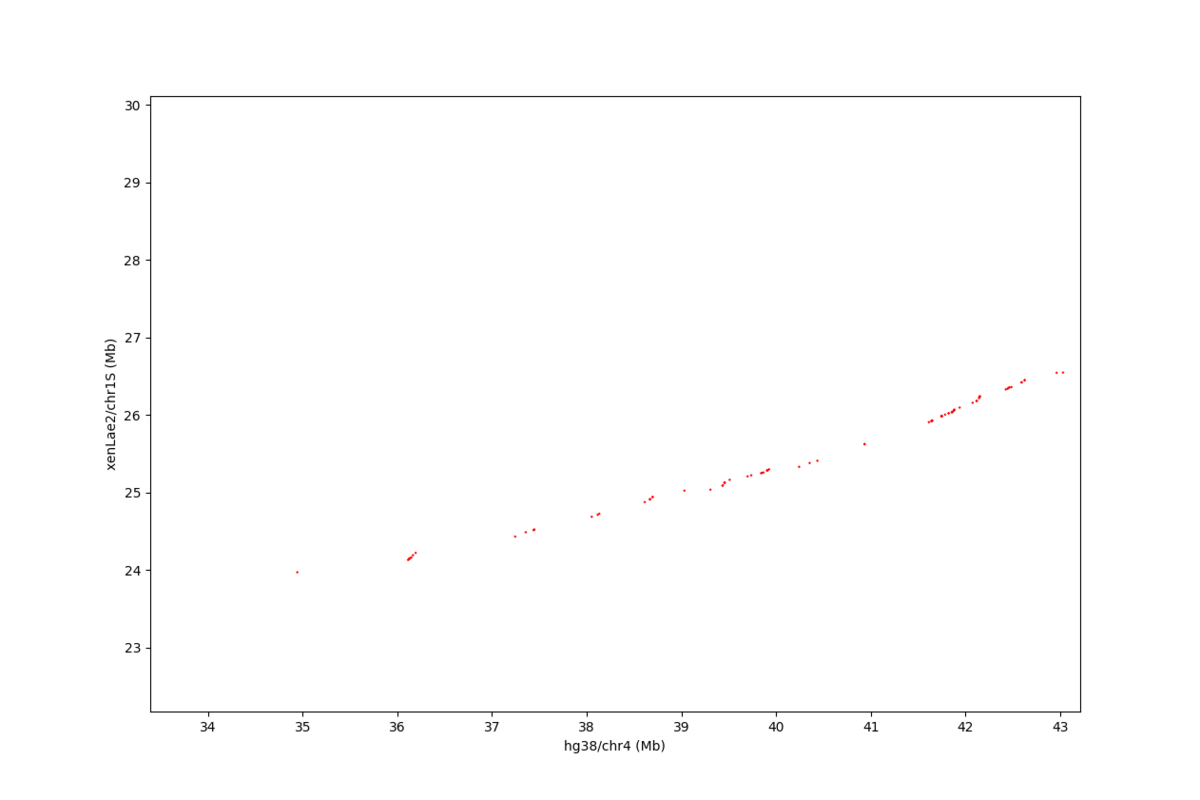
数字で見ると、ヒトとアフリカツメガエルは、それほど似てないけど、ドットプロットを見ると、マッピングされた領域が、かなり広い範囲で一直線上に綺麗に並んでることが、割とある。以下は、ヒト4番染色体とアフリカツメガメル1S染色体のドットプロット
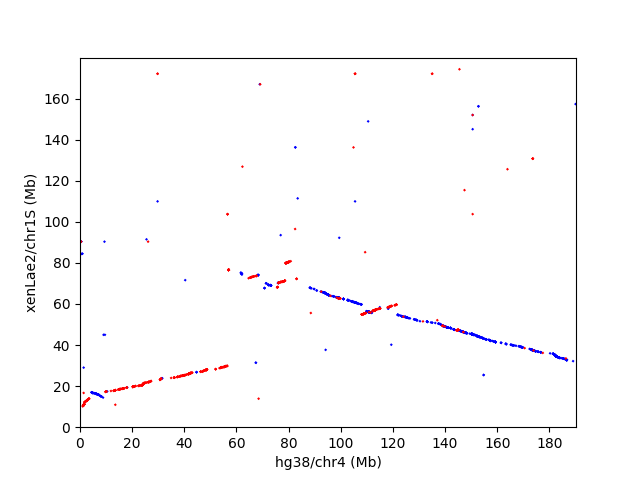
画像で見ると、凄く密に相同配列がありそうに見えるけど、これは可視化の仕方の問題で、拡大すると、相同配列は、もっと、まばらにしかないことが分かる。以下は、上のドットプロットの一部を拡大したもの。
ヒトと魚くらいになると、そのような領域は、もっと断片的にしか存在しない。もっと遠い種との間でも、遺伝子の並び順は保存されてたりするらしく、このことを指してシンテニーとか呼ぶ(Hox geneクラスターみたいなもん?)。シンテニーの解析から、脊索動物の系統関係を推測したりしてる論文もある。
The amphioxus genome and the evolution of the chordate karyotype
https://doi.org/10.1038/nature06967